Ulcus cruris, Antiphospholipidsyndrom, Marcumar
-
Schwierigkeitsgrad Fortgeschrittene
47 Ulcus cruris bei Antiphospholipidsyndrom unter Marcumar-Therapie
Last Updated: 20-08-2013
Nina van Beek¹, Nina Schumacher¹, Ozan Haase¹, Detlef Zillikens¹, Birgit Kahle¹, Enno Schmidt¹,²
1 Klinik für Dermatologie, Allergologie und Venerologie, Universität zu Lübeck
2 Exzellenzzentrum für Entzündungsmedizin, Universität zu Lübeck
vorgestellt als Dia-Klinik, 46. Tagung der Deutschen Dermatologischen Gesellschaft, Dresden 2013
DOI
10.1111/ddg.12061
Anamnese
Bei einem 72-jährigen Patienten trat ein innerhalb von 6 Wochen rasch größenprogredientes, schmerzloses Ulcus an der rechten Wade auf. In einer Probebiopsie zeigten sich Regeneratepithel mit darunter liegenden Gefäßproliferaten. Eine Behandlung mit verschiedenen Externa, Wundauflagen und Kompressionstherapie mit Kompressionsstrümpfen der Klasse II konnte keine Besserung erzielen. Aufgrund einer Tachyarrhythmia absoluta bei Vorhofflimmmern erfolgt seit Jahren eine Therapie mit Phenprocoumon (Marcumar®). Unter Antikoagulation war ein Jahr zuvor bereits ein Apoplex aufgetreten. Als weitere Vorerkrankungen waren u. a. ein Diabetes mellitus Typ 2 (medikamentös eingestellt), eine monoklonale Gammopathie unklarer Signifikanz sowie Z. n. Kolon- und Oropharynxkarzinom bekannt. Raynaudsymtomatik bestand nicht.
Hautbefund
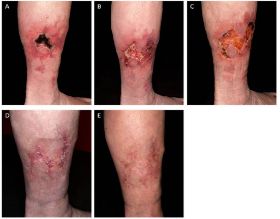
An der rechten Wade ein 5 x 3 cm großes, bizarr konfiguriertes Ulcus mit zentraler trockener Nekrose. In der Umgebung angedeutet retikuläres düsterrotes Erythem (Abb. 1). Die übrige Haut war intakt.
Dermatohistopathologie
Unauffälliges Epithel mit korbgeflechtartiger orthologer Verhornung. Im Fettgewebe reichlich deutlich lumenverengte und verkalkte arterielle Blutgefäße. Keine Vaskulitis. Keine Nekrose.
Diagnose: Ausgeprägte lumenstenosierter verkalkender Arteriosklerose mittelgroßer Arterien im Fettgewebe. Kein Hinweis auf eine Vaskulitis oder Marcumar-Nekrose.
Weitere Diagnostik
Labor initial (auszugsweise): INR 2,76, Quick-Wert 23 %, Thrombozyten 225/nl (150-400/nl), HbA1c 6,5 % Hb (4,3-6,1 % Hb)
Gefäßdiagnostik: Dopplersonographisch zeigte sich kein Hinweis für eine CVI oder pAVK.
Gerinnungsdiagnostik: Nachweis einer heterozygoten MTHFR (Methyltetrahydrofolsäure-Reduktase)-Mutation.
Andere Koagulopathien wurden serologisch, eine Vaskulitis histologisch und mittels direkter Immunfluoreszenz ausgeschlossen.
Autoimmundiagnostik:
Cardiolipin-AK (IgG): 37,6 <10 U/ml
β2-Glykoprotein I-AK: 24,7 <10 U/ml
Phosphatidylserin-AK (IgG und IgM): 33,1 <10 U/ml
Antinukleäre Antikörper (ANA) Screen i. S.: negativ
pANCA/cANCA i. S.: negativ
Komplement C3/C4, Rheumafaktor, Anti-CCP i. S.: normwertig
Kryoglobuline/Kryofibrinogen/Kälteagglutinine: negativ
Immunelektrophorese: freie Lambda-Leichtketten
Therapie und Verlauf
Allgemein
Nach Vorliegen der ausführlichen Gerinnungs- und Autoimmunitätsdiagnostik zeigten sich Autoantikörper gegen Cardiolipin, β2-Glykoprotein I und Phosphatidylserin. Zudem fiel die monoklonale Gammopathie unklarer Signifikanz mit Erhöhung der Leichtketten in Serum und Urin auf. Im Ganzkörper-CT ergab sich kein Hinweis auf ein Plasmozytom. In Zusammenschau der Klinik und aller Befunde stellten wir die Diagnose eines primären Antiphospholipidsyndroms (APL-Syndrom).
Therapeutisch wurde die Antikoagulationstherapie auf Enoxaparin (Clexane®) 0,8 mg s.c. 2x/d umgestellt und eine immunsuppressive Therapie mit Dexamethason 100 mg/d i.v. über 3 aufeinander folgende Tage und Azathioprin 200 mg/d per os eingeleitet. Insgesamt erfolgten 6 Zyklen Dexamethason-Pulstherapie über 5 Monate. Die Lokaltherapie erfolgte mittels stadiengerechten Wundauflagen sowie Kompression mittels Kompressionsstrümpfen der Klasse II. Unter dieser Therapie konnte innerhalb von 23 Wochen eine vollständige Abheilung des Ulcus beobachtet werden. Die Medikation mit Azathioprin konnte ab Woche 9 reduziert und nach 65 Wochen abgesetzt werden. Clexane® wurde nach 25 Wochen wieder auf Marcumar® umgesetzt. Nach einer Nachbeobachtungszeit von 26 Monaten war es zu keinen neuen Ulzera oder einer weiteren thromboembolischen Komplikation gekommen.
Kommentar
Das APL-Syndrom ist durch rezidivierende venöse und arterielle Thrombosen, einhergehend mit erhöhten Antikörpern gegen Phospholipide gekennzeichnet. Es tritt vorwiegend bei jungen Frauen auf. Dem primären Antiphospholipidsyndrom liegen keine anderen Erkrankungen zugrunde, das sekundäre Antiphospholipidsyndrom tritt in Assoziation vor allem mit dem systemischen Lupus erythematodes auf, aber auch mit anderen Autoimmunerkrankungen oder Infektionskrankheiten. Die Klinik des APL-Syndroms ist vielfältig und imponiert z. B. als transitorisch ischämische Attacken, Myokardinfarkt, Nierenversagen sowie bei weiblichen Patienten durch habituelle Aborte. Kutane Manifestationen äußern sich in Raynaud-Symptomatik, Livedo racemosa, Petechien und schmerzhaften, akral betonten Hautnekrosen. Hämatologisch zeigt sich nicht selten eine Thrombozytopenie. Der Verlauf ist meist subakut, selten akut-lebensbedrohlich in Form eines "catastrophic" APL-Syndroms mit thrombotischen Ereignissen an mehreren Organen. Als Pathomechanismen der thrombophilen Diathese werden unter anderem die direkte Endothelzellschädigung, die Hemmung der Protein-C-Aktivierung und die Bindung von β2-Glykoprotein-I sowie die Hemmung der endothelialen Stickstoffmonoxid-Synthethase diskutiert. Die Diagnose wird gemäß den Sapporo-Kriterien gestellt.
| Klinische Kriterien | Gefäßverschlüsse (≥1 klinisches Ereignis einer arteriellen, venösen oder "Small-Vessel"-Thrombose) |
| Schwangerschaftskomplikationen (≥1 Abort in/nach der 10. SSW oder ≥1 Frühgeburt in/vor der 34. SSW oder ≥ 3 Aborte (konsekutiv) vor der 10. SSW) | |
| Laborkriterien (jeweils bei ≥2 Messungen in mindestens 12 Wochen Abstand) | Antikardiolipin-Antikörpern vom IgG- oder IgM-Subtyp in mittlerem oder hohem Titer |
| Nachweis von Lupusantikoagulans | |
| Anti-β2-Glykoprotein-1-Antikörper vom IgG- und/oder IgM-Typ |
Ein definitives APL-Syndrom liegt vor, wenn mindestens ein klinisches und ein Laborkriterium erfüllt sind.
Die Therapie des APL-Syndroms erfolgt mit oraler Antikoagulation. Jedoch wurden thrombotische Ereignisse bei Patienten mit APL-Syndrom auch unter adäquater Antikoagulation beschrieben. Zusätzlich kommen (besonders bei akutem Verlauf) hochdosierte Kortikosteroide evtl. in Kombination mit Immunsuppressiva zum Einsatz. Weitere Therapieansätze sind Hydroxychloroquin, Statine, Rituximab und IVIG. Ulcera crurum im Rahmen eines APL-Syndrom werden auch als Form eines Pyoderma gangraenosum interpretiert. Bei sekundärem APL-Syndrom erfolgt zudem die Therapie der Grunderkrankung.
Unser Patient entwickelte unter einer bestehenden Marcumartherapie ein therapierefraktäres Ulcus cruris mit Nekrose. Es konnten erhöhte Antikardiolipinund Anti-β2-Glykoprotein-I-Antikörper nachgewiesen werden. Retrospektiv ist als weitere klinische Manifestation des APL-Syndroms der Apoplex zu bewerten. Unter Therapieintensivierung mit hochdosierten Glukokortikosteroiden als i.v. Dexamethason-Pulstherapie in Kombinantion mit Azathioprin und Enoxaparin s.c. konnte ein Abheilen des Ulcus erzielt werden. Diese antiinflammatorische Kombinationsbehandlung aus s.c. niedermolekularem Heparin, Dexamethason i.v. Pulsen und Azathioprin wurde nebenwirkungsfrei vertragen und induzierte eine lang anhaltende Remission. Dieser Fall zeigt die Bedeutung der differenzialdiagnostischen Abklärung atypischer Befundkonstellationen beim Ulcus cruris, bei welcher ein APL-Syndrom in Betracht gezogen werden sollte. Zudem stellt die Marcumarisierung keine sichere Prophylaxe der kutanen Manifestationen eines APL-Syndroms dar.
Literatur
1 Bordin G, Boldorini R, Meroni PL. The two hit hypothesis in the antiphospholipid syndrome: acute ischaemic heart involvement after valvular replacement despite anticoagulation in a patient with secondary APS. Lupus 2003; 12: 851-853.
2 Canas CA, Duran CE, Bravo JC, Castano DE, Tobon GJ. Leg ulcers in the antiphospholipid syndrome may be considered as a form of pyoderma gangrenosum and they respond favorably to treatment with immunosuppression and anticoagulation. Rheumatol Int 2010; 30: 1253-1257.
3 Erkan D, Lockshin MD. New approaches for managing antiphospholipid syndrome. Nat Clin Pract Rheumatol 2009; 5: 160-170.
4 Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, Derksen RH, PG DEG, Koike T, Meroni PL, Reber G, Shoenfeld Y, Tincani A, Vlachoyiannopoulos PG, Krilis SA. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4: 295-306.
5 Pengo V, Ruffatti A, Iliceto S. The diagnosis of the antiphospholipid syndrome. Pathophysiol Haemost Thromb 2006; 35: 175-180.
6 Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, Brey R, Crowther M, Derksen R, Erkan D, Krilis S, Machin S, Pengo V, Pierangeli S, Tektonidou M, Khamashta M. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: report of a task force at the 13th International Congress on antiphospholipid antibodies. Lupus 2010; 20: 206-218.