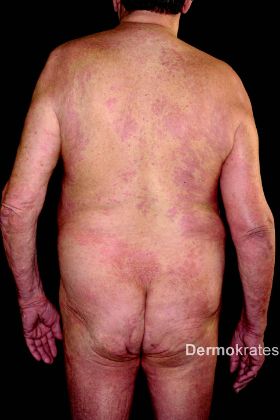
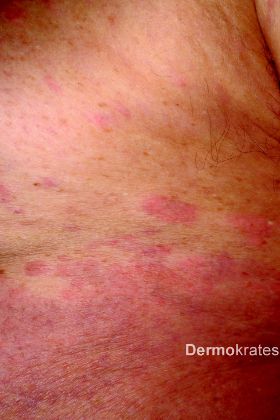
Ein 74-jähriger Patient stellt sich mit stark pruriginösen Hautveränderungen vor. Diese seien erstmals während einer fünf Monate zurückliegenden Urlaubsreise nach Mauritius aufgetreten und hätten seither stetig zugenommen. Ferner bestünden Arthralgien im Bereich der Fingerund Sprunggelenke. Ein ambulanter Therapieversuch mit Antihistaminika habe nur wenig Besserung erbracht.
-
Schwierigkeitsgrad Fortgeschrittene
26 Myeloproliferatives Hypereosinophilie-Syndrom
Ilana Goldscheider1, Attila S. Antal1, Michael J. Flaig1, Thomas Herzinger1, Ralf Schmidmaier2, Thomas Ruzicka1
1 Klinik und Poliklinik für Dermatologie und Allergologie der Ludwig-Maximilians-Universität München
2 Medizinische Klinik Innenstadt des Klinikums der Universität München
vorgestellt als Dia-Klinik, 23. Fortbildungswoche für praktische Dermatologie und Venerologie, München 2012
DOI
Histopathologie
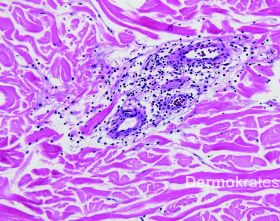
Rücken links: Insbesondere oberflächlich perivaskulär, in geringem Maß auch tief perivaskulär gemischtzelliges entzündliches Infiltrat, das zum Einen aus Lymphozyten, zum Anderen aus eosinophilen Granulozyten besteht. Intravasal fast ausschließlich eosinophile Granulozyten. Interstitiell deutlich weniger eosinophiles Infiltrat. Auf den vorliegenden Schnittstufen kein Nachweis von Eosinophilengranula in der Umgebung kollagener Fasern der Dermis ("Flammenfiguren"). Der Befund ist gut vereinbar mit einem Hypereosinophilie-Syndrom.
Labor
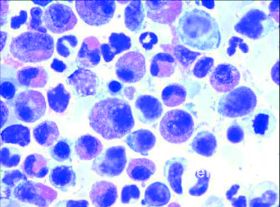
Hämoglobin mit 12,2 g/dl [14,0-17,5] erniedrigt, Thrombozyten mit 129,0 10³/μl [150,0-300,0] erniedrigt,
Leukozyten mit 23,0 10³/μl [4,0-11,3] erhöht, im Differenzialblutbild ausgeprägte Eosinophilie bis 70 % [1-4], Eosinophile absolut mit 16100/μl [50-250] stark erhöht, ein Blast und ein Metamyelozyt; LDH mit 334 U/l [<250], CRP mit 2,75 mg/dl [<0,50], Troponin-T mit 0,084 ng/ml [<0,014] und Gesamt-IgE mit 1294 kU/l [<100 kU/l] erhöht.
Liquorzytologie mit Immunphänotypisierung
Die Zellzahl ist im Normbereich, das Zellbild ist lymphozytär mit Eosinophilie. Letztere bei entsprechender klinischer Symptomatik dringend verdächtig auf ZNS-Beteiligung im Rahmen des Hypereosinophilie-Syndroms.
Stuhlparasitologie
Keine Wurmeier, Lamblien, Amöben oder Kryptosporidien.
Knochenmarkspunktion
Histologie: Deutlich hyperzelluläres Beckenkammtrepanat mit Infiltraten einer reifzelligen myeloproliferativen Neoplasie mit massiver Eosinophilie ohne Vermehrung und Atypien der Mastzelllinie. Eine begleitende systemische Mastozytose liegt somit nicht vor.
Zytomorphologie: Vereinbar mit einem Myeloproliferativen Syndrom.
Immunphänotypisierung: Immunzytologisch kein Anhalt für akute Leukämie, T-NHL oder Mastozytose. Ausgeprägte Eosinophilie aber keine Expression von CD117, CD34. Myeloblasten nicht vermehrt.
Molekularbiologie: Nachweis einer JAK2-V617FMutation.
Zytologie des peripheren Blutes
Ausgeprägte Eosinophilie (57 %). Die Eosinophilen sind überwiegend ausgereift.
Immunphänotypisierung aus dem peripheren Blut
Kein Anhalt für eine akute Leukämie. Ausgeprägte Eosinophilie (70 %).
Molekularbiologie des peripheren Blutes
Nachweis einer JAK2-V617F-Mutation. Kein Nachweis des FIP1L1-PDGFRalpha-Fusionstranskriptes mittels PCR und FISH.
Apparative Diagnostik
Elektrokardiogramm: Herzfrequenz 83/min. Leichte ST-Streckensenkung.
Herzkatheteruntersuchung: Diffuse koronare Herzerkrankung ohne hämodynamisch relevante Stenosen.
Sonographie des Abdomen: Leichte Splenomegalie.
Röntgen des Thorax: Zeichen des (vorbekannten) Lungenemphysems.
Neurologisches Konsil
Deutliche Enzephalopathie mit wohl hirnorganischem Psychosyndrom. Rechtsseitige Halbseitensymptomatik ohne Korrelation im cranialen CT. Verdacht auf Polyneuropathie. EEG mit Hinweis auf mittelschwere Enzephalopathie.
Therapie und Verlauf
Allgemein
Im Laufe des stationären Aufenthaltes klagt der Patient über neu aufgetretenen Schwindel und Sehstörungen. Am Folgetag klagte er bei der Morgenvisite über plötzlich aufgetretenen retrosternalen Schmerz. Aufgrund der Konstellation von pruriginösem makulopapulösem, teils urtikariellem Exanthem, ausgeprägter Blut- und Gewebseosinophilie sowie dem Auftreten neurologischer (Schwindel, Verschwommensehen) und kardialer Symptome (retrosternaler Schmerz, EKG-Veränderung, Troponin-T-Erhöhung) konnte die Verdachtsdiagnose eines Hypereosinophilie-Syndromes (HES) gestellt werden. Bei vital gefährdender neurologischer und kardialer Beteiligung erfolgte eine Verlegung in die Medizinische Klinik Innenstadt der LMU. Im Verlauf kam es zu einer dramatischen Entwicklung der neurologischen Symptome mit rasch progredienter Enzephalopathie und Halbseitensymptomatik, so dass der Patient kaum noch erweckbar war. Nach Einleitung einer hochdosierten Glukokortikosteroidtherapie (1 mg/kg Körpergewicht Prednisolon) unter Komedikation mit Imatinib 400 mg p.o. täglich zeigten sich die neurologischen, kardiologischen und dermatologischen Symptome rasch rückläufig. Die Eosinophilen zeigten sich mit um 17 % stabil. Bei fehlendem Nachweis des FIP1L1-PDGFRalpha-Fusionstranskriptes wurde Imatinib sistiert und stattdessen eine Therapie mit Hydroxycarbamid 1000 mg 2 x täglich eingeleitet. Prednisolon konnte bis zur Entlassung des Patienten auf 5 mg täglich reduziert werden. Eine spezifische Antikörpertherapie mit einem JAK2-Inhibitor wird aufgrund des Nachweises einer JAK2-V617F-Mutation von den behandelnden Kollegen der Abteilung für Hämato-Onkologie erwogen.
Kommentar
Das Hypereosinophilie-Syndrom (HES) mit seinen Subtypen idiopathisches HES, myeloproliferatives HES, lymphozytisches HES, familiäres HES und HES-Overlap-Syndrom, bezeichnet eine heterogene Gruppe von Erkrankungen, die durch eine überschießende Produktion eosinophiler Granulozyten mit konsekutiver Infiltration und Schädigung verschiedener Organe gekennzeichnet ist. Liegt die absolute Zahl der eosinophilen Granulozyten im peripheren Blut zweimalig über 1500/μl und finden sich keine anderen plausiblen Erklärungsansätze für die vorliegende Eosinophilie, wie zum Beispiel eine Parasitose, so kann nach neuerer Definition vom Vorliegen eines HES ausgegangen werden.
Genauere epidemiologische Daten zur Prävalenz des HES liegen nicht vor, es handelt sich jedoch um ein sehr seltenes Krankheitsbild. Die Pathogenese des HES ist nicht geklärt. Es wird vermutet, dass die Überproduktion von Eosinophilen im Knochenmark auf einer Überstimulation durch das die Eosinophilopoese fördernde Zytokin IL-5, einer klonale Proliferation von hämatopoetischen Stammzellen oder einer Fehlregulation anderer noch nicht bekannter Einflussfaktoren beruht.
Die myeloproliferative Variante des HES ist durch das Vorhandensein von Symptomen myeloproliferativer Erkrankungen wie Hepato- und/oder Splenomegalie, Anämie, Thrombozytopenie, dem Vorkommen von Blasten im peripheren Blut und dem Nachweis von Chromosomenabnormalitäten gekennzeichnet. Ein Übergang in eine chronische eosinophile Leukämie (CEL) ist möglich. Bei Nachweis einer Translokation im FIP1L1-PDGFRalpha-Gen auf Chromosom 4q12 ist daher analog der chronisch eosinophilen oder der chronisch myeloischen Leukämie ein gutes Ansprechen auf Tyrosinkinasinhibitoren wie Imatinib zu beobachten. Durch die Unterbrechung der Dauerstimulation durch die PDGFR-Tyrosinkinase wird der Proliferationsreiz auf die eosinophilen Vorläuferzellen unterbrochen. Bei unserem Patienten war diese Mutation nicht nachweisbar. Allerdings konnte mittels PCR sowohl im peripheren Blut als auch im Knochenmark ein kleiner Klon mit einer JAK2-V616F-Mutation identifiziert werden. Es handelt sich hierbei um eine Mutation einer "Janus-like-Kinase", die bei 30-95 % aller chronischen myeloproliferativen Neoplasmen (Chronisch myeloische Leukämie, chronische eosinophile Leukämie, Polycythämia vera, essentielle Thrombozythämie, Myelofibrose) nachgewiesen werden kann. Verschiedene JAK2-Inhibitoren werden zurzeit in klinischen Studien getestet.
Die Erstlinientherapie des HES besteht bei fehlendem Mutationsnachweis oder mangelnder Verfügbarkeit von mutationsspezifischen Antikörpern aus einer hochdosierten Glukokortikosteroidtherapie. Als steroidsparende Medikamente werden Hydroxyurea, Interferon-alpha und im Rahmen von klinischen Studien der Anti-Interleukin-5-Antikörper Mepolizumab eingesetzt.
Die klinische Präsentation des HES ist sehr variabel. Die Erstkonsultation und Erstdiagnose erfolgt allerdings häufig durch den Dermatologen, da Hautveränderungen in über einem Drittel der Fälle auftreten. Diese manifestieren sich als pruriginöse, makulopapulöse bis urtikarielle Exantheme, in seltenen Fällen auch als Erythrodermie oder im Rahmen der Organschädigung als digitale Nekrosen. Eine Organschädigung durch die toxischen Produkte der Eosinophilengranula (wie eosinophiles kationisches Protein, eosinophile Peroxidase oder Major Basic Protein) können den Gastrointestinaltrakt, das ZNS, die Lunge und das Herz betreffen. Mortalität und Morbidität sind vor allem durch eine kardiale Beteiligung bedingt. Diese tritt in ca. 5 % aller HES auf, allerdings scheint sie bei der myeloproliferativen Variante höher zu liegen. In der akuten Phase kommt es dabei zu einer in der Regel klinisch stummen nekrotisierenden Endo- und Myokardschädigung. In der Folge kann es zur Bildung von Thromben und als gravierende Komplikation zu Thrombembolien kommen. Eine zerebrale Thrombembolie liegt wahrscheinlich auch den oben genannten neurologischen Komplikationen unseres Patienten zugrunde. Bei nachgewiesener Liquoreosinophilie ist aber auch von einer zusätzlichen direkten toxischen ZNS-Schädigung auszugehen.
Fazit
Dieser Fall ist von besonderem Interesse für den Dermatologen, da eine Hautbeteiligung als Erstmanifestation dem lebensbedrohlichen Verlauf mit Multiorganbeteiligung um Monate vorausging. In der Zusammenschau von Hautbefund, Blut- und Gewebseosinophilie, und in Kenntnis möglicher Komplikationen konnte zügig eine Therapie unter engmaschiger Kontrolle der Vitalparameter eingeleitet werden. Trotz seiner Seltenheit ist das HES aufgrund möglicher schwerer Verlaufsformen eine wichtige Differenzialdiagnose bei mit Blut- und Gewebseosinophilie einhergehenden Hautveränderungen.
Literatur
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, et al. (2003) A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 348: 1201-1214.
Grunewald SM, Bröcker ES (2005) Dermatologische Erkrankungen mit eosinophilen Granulozyten. In: Plewig G, Kaudewitz P, Sander CA (Hrsg.) Fortschritte der praktischen Dermatologie und Venerologie. Bd. 19, Springer, Heidelberg, S 73-86.
Roufosse F, Klion AD, Weller PF (2011) Clinical manifestations, pathophysiology, and diagnosis of the hypereosinophilic syndromes. www.uptodate.com
Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, et al. (2010) Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 126: 45-49.