Eine 62-jährige Patientin stellte sich mit einer Überweisung aus einer Venenklinik mit seit einigen Wochen bestehenden Schwellungen beider Beine und knotigen Veränderungen im Bereich des oberflächlichen Venensystems beidseits vor. Es sei bereits eine Therapie bei Verdacht auf eine Thrombophlebitis durchgeführt worden, diese hätte jedoch keine Besserung der Beschwerdesymptomatik erbracht. Zusätzlich habe die Patientin gewollt 15 kg in 6 Monaten abgenommen. Weitere B-Symptomatik bestünde nicht.
-
Schwierigkeitsgrad Fortgeschrittene
5 Intravaskuläres großzelliges B-Zell-Lymphom
Sebastian Ziegler, Rudolf Stadler
Dermatologie, Johannes Wesling Klinikum Minden
vorgestellt als Dia-Klinik, 46. DDG-Tagung, Dresden 2011
DOI
Hautbefund
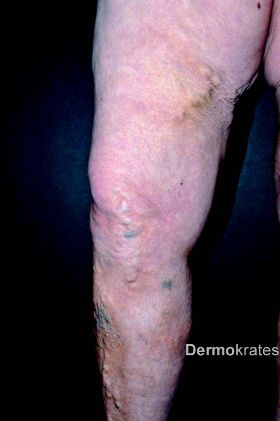
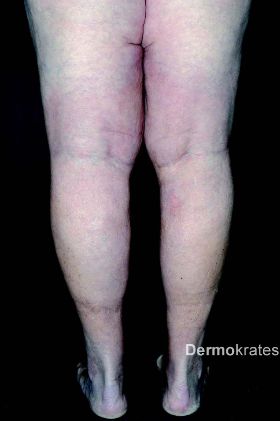
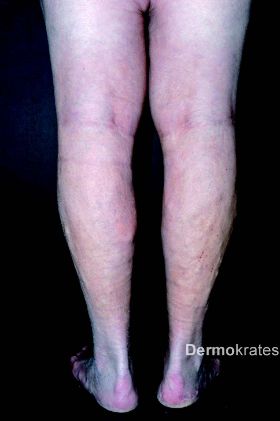
Es zeigen sich beidseits an den Oberschenkeln medial bis über das Knie ziehend im Verlauf der Vena saphena magna derbe, indurierte Areale, teils knotig verändert, jedoch keine epidermale Beteiligung. Die Hautveränderungen zeigen sich wenig bis gar nicht druckdolent. Im Bereich beider Unterschenkel imponieren tastbare, subcutan liegende Knoten ohne epidermale Beteiligung neben ektetisch erweiterten Venen. Beide Beine sind leicht geschwollen. Weiterhin zeigt sich eine diffuse Alopezie am Capillitium. Keine vergrößerten Lymphknoten tastbar.
Labor
LDH 923 U/l (135-225), Hb 101 g/l (120-160), D-Dimer 3,25 ug/dl (<0,5), ANA 1:1280 (<1:80), Anticardiolipin-Ak IgG 56,30 (<12), C-ANCA pos. (neg.), P-ANCA pos. (neg.), Protein 3 quant. 78,5 u/ml (<7), Proteinase 3 pos. (neg.), MPO pos. (neg.), Kathesin G pos. (neg.), MPO-AK quant. 56 (<7), C4-Komplement 144 mg/l (180-490), fT3 1,3 ng/l (2,0-4,4), fT4 6,4 ng/l (9-19)
Eiweiß-Elektrophorese: Albumin 40,2 % (59,0-70,6), Gamma-Glob. 39,6 % (11,2-19,9), IgG im Serum 32,60 G/L (7,0-16,0), Kappa i. Serum 4,90 G/L (1,7-3,7), Lambda i. Serum 5,40 G/L (0,92-2,1), Kappa/Lambda Quotient 0,91 (1,35-2,65)
Unauffällig waren: übriges Routine-Labor, DNS-Ak, Anticard. IgM, C3-Komplement, Procalcitonin, CHE, Gerinnung, Kryoglobuline, Kälteagglutinine, MAK, TAK, TRAK
Dermatohistopathologie
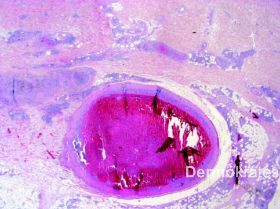
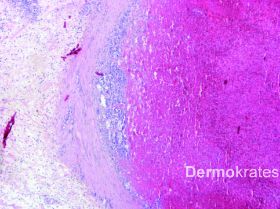
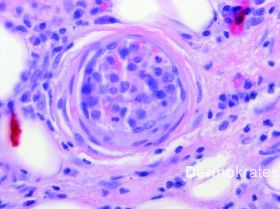
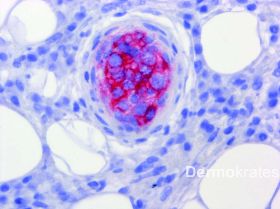
Es zeigen sich in der HE-Färbung intravasal gelegene pleomorphe B-Zell-Lymphozyten mit runden oder ovalen chromatindichten Kernen neben einem das gesamte Gefäß ausfüllenden fibrinösen Thrombus. Immunhistologisch stellen sich die Tumorzellen innerhalb des Lumens CD20, CD79a, und MUM1 positiv dar, verhalten sich negativ gegenüber CD10, ALK-1 und CD3. Mit dem Proliferationsmarker MiB zeigt sich im Lumen eine extrem hohe Proliferationsrate von 70-80 %. Die einzelnen B-Zellen exprimieren im Lumen ausgeprägt BcL-2. BcL-6 wird innerhalb des Lumens im Wesentlichen nicht exprimiert.
Molekularpathologie
Nachweis einer klonalen B-Zell-Lymphozytenpopulation mit der Vκ-Kde Region mit zwei Basenpaaren 279 und 284. Ein monoklonales Muster der Immunglobulinschwerkette konnte nicht nachgewiesen werden.
Apparative Diagnostik
Duplexsonographie des Venensystems: Insuffizienz Vena saphena magna bds. II-III°, Thrombophlebitis SA der VSM rechts, tiefes Venensystem frei
Lymphknotensonographie: unauffällig
Knochenmarkstanze: keine Infiltration durch das B-Zell-Lymphom
MR-Schädel: unauffällig
PET-CT: viszerale Organe und Lymphknoten unauffällig, gesteigerte Glucoseutilisation distale Femora, proximale Tibia rechts, BWK 4, LWK 1, Kreuzbein
Therapie und Verlauf
Allgemein
Unter dem klinischen Bild einer Thrombophlebitis erfolgte bei Therapieresistenz und ungewöhnlicher Verteilung die Entnahme einer Probebiopsie. Hierbei wurde histologisch, immunhistologisch und molekularpathologisch ein intravaskuläres großzelliges B-Zell-Lymphom diagnostiziert (T3N0M0). Auch gelang der Nachweis einer klonalen Lymphozytenpopulation. In der weiteren Durchuntersuchung zeigten sich keine Hinweise für eine Lymphknoten- oder Organbeteiligung. Nach Empfehlungen der hausinternen interdisziplinären Hauttumorkonferenz wurde bei der Patientin eine Therapie nach dem R-CHOP-Protokoll (Rituximab, Cyclophosphamid, Hydroxy-Doxorubicin, Vincristin, Prednisolon) eingeleitet. Zusätzlich erhielt die Patientin Enoxiparin 2x80 mg am Tag sowie konsequente Kompressionstherapie beider Beine. Im Verlauf rasche Besserung der Beschwerdesymptomatik sowie des klinischen Befundes.
Im erneuten Staging nach 6 Zyklen R-CHOP Chemotherapie zeigte sich klinisch sowie apparativ kein Lymphomnachweis. Der weitere klinische Verlauf bleibt abzuwarten.
Zu diskutieren bleibt, ob die bei unserer Patientin beobachteten Autoimmun-Befunde einer eigenen Krankheitsentität zuzuordnen sind oder ob es durch das B-Zell-Lymphom zu Autoimmunprozessen kommt, wie sie zum Beispiel beim angioimmunoblastischen Lymphom beschrieben sind. Auch hier werden Verlaufskontrollen erfolgen.
Kommentar
Das intravaskuläre B-Zell-Lymphom (IVL) ist eine sehr seltene Erkrankung, die mit Proliferation multizentrisch auftretender intravaskulärer Tumorzellinfiltrate zu einer Okklusion kleiner und mittlerer Gefäße der Organe führt. Typischerweise sind die Patienten älter (>60 Jahre), Männer und Frauen erkranken gleich häufig. Je nach Muster des Organbefalls ergibt sich eine heterogene klinische Symptomatik. Bei ca. 39 % der Patienten treten heterogene neurologische Symptome bei zerebraler Beteiligung auf. Ein MRT des Schädels sollte im Rahmen der Staginguntersuchungen durchgeführt werden. Die Veränderungen sind jedoch unspezifisch und ähneln insgesamt einer Vaskulitis, sie lassen sich zu Lebzeiten nur bei ca. 50 % der Patienten nachweisen. Auch der Einsatz eines PET-CT hat sich in einzelnen Fallberichten als wertvolle Ergänzung der Diagnostik erwiesen. Die sehr seltene isoliert kutane Variante, wie bei unserem Patienten beschrieben, wird in der aktuellen WHO-EORTC-Klassifikation der kutanen B-Zell-Lymphome als eigene Entität aufgeführt. Sie hat einen eher günstigen Verlauf mit bekannt gutem Therapieansprechen. Die 3-Jahres-Überlebensrate wird mit 56 % angegeben. Die Diagnose des intravaskulären B-Zell-Lymphoms wird histologisch, immunhistologisch und molekularpathologisch gestellt.
Die Behandlung erfolgt entsprechend den Protokollen nodaler B-Zell-Lymphome. Randomisierte, kontrollierte Therapiestudien zur Behandlung intravaskulärer B-Zell-Lymphome liegen bisher nicht vor.
Literatur
Ferri AJ, Campo E, Seymour JF et al. (2004) Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with spezial emphasis on the ”cutaneous variant”. Br J Haematol 127: 173-183.
Olsen E, Vonderheid E, Pimpinelli N et al. (2007) Revision to the staging and classification of mucosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force European Organisation of Research and Treatment of Cancer (EORTC). Blood 110: 1713-1722.
Willemze R, Jaffe ES, Burg G et al. (2005) WHO-EORTC classification for cutaneous lymphomas. Blood 105: 3768-3785.
Yasemin S, Cuneyt T, Bülent S et al. (2010) 18F-FDG PET/CT in a case of intravascular large B-cell-lymphoma. Eur J Nucl Med Mol Imaging 37:1801.