In Begleitung der leiblichen Mutter stellt sich ein 8 Monate altes Mädchen mit verrukösen Hautveränderungen betont an der oberen und unteren Extremität vor. Die Läsionen bestünden seit der zweiten Lebenswoche und seien initial zunächst makulös sowie teils vesiko-bullös in Erscheinung getreten. Im Verlauf der ersten Lebensmonate sei es zum Auftreten von verrukösen Plaques gekommen. Aktuell verschlechtere sich der Hautbefund progredient. Bei der Patientin war bereits pränatal durch Chorionzottenbiopsie die Diagnose einer Incontinentia pigmenti gestellt worden. An systemischen Grunderkrankungen bestehen weiterhin eine primäre pulmonale Hypertonie mit Sauerstoff-Substitutionspflichtigkeit, eine symptomatische Enzephalopathie, ein Mikrozephalus, sowie rezidivierende Netzhautblutungen. Die Patientin befand sich postpartal in längerfristiger intensivmedizinischer Betreuung. Auch bei der Mutter der Patientin war bereits im frühen Erwachsenenalter die molekulargenetisch gesicherte Diagnose "Incontinentia pigmenti" (Stopmutation in Codon 265 im IKBKG [alias NEMO]-Gen) gestellt worden. Systemische Grunderkrankungen seien bei der Mutter nicht bekannt, und nach nicht näher eruierbaren Hautveränderungen in der Kindheit bestehen bei ihr aktuell nur Nageldystrophien sowie Paronychien an den proximalen Nagelfalzen beider Zeigefinger.
-
Schwierigkeitsgrad Fortgeschrittene
31 Incontinentia pigmenti (Bloch-Sulzberger-Syndrom)
Till Geimer, Julia Walch, Ilana Goldscheider, Kathrin Giehl
Klinik und Poliklinik für Dermatologie und Allergologie der Ludwig-Maximilians-Universität München
vorgestellt als Dia-Klinik, 23. Fortbildungswoche für praktische Dermatologie und Venerologie, München 2012
Hautbefund
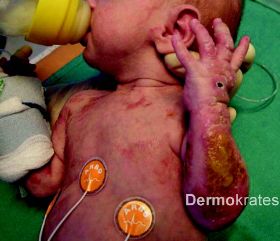
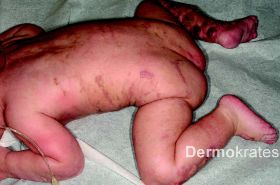
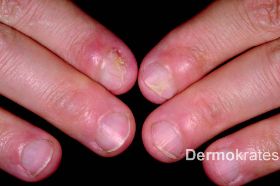
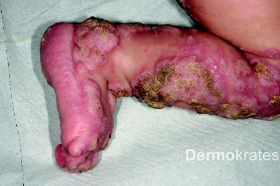
An der Stirn rautenförmige 1,5 cm große atrophe, hautfarbende Makula. Nuchal zahlreiche bis zu 5 mm große erythematöse Makulae, teils flächig konfluierend. Am Stamm und den Extremitäten proximal disseminiert streifige bis wirbelförmige hellbraune Hyperpigmentierungen die eine Verteilung entlang der Blaschko-Linien aufweisen. Am rechten Zeigefinger sowie am rechten Handgelenk beugeseitig mehrere bis zu 8 mm große rot-braune Knoten mit verruköser Oberfläche. An der unteren Extremität distal betont multiple erythematöse, scharf begrenzte, bis 1,2 cm große Knoten, die zu großflächigen Plaques mit bräunlich-hyperkeratotischer, teils verruköser Oberfläche konfluieren.
Therapie und Verlauf
Allgemein
Neben regelmäßigen Ölbädern kam prednicarbat-/polyhexanidhaltige Creme 2 mal täglich am betroffenen Integument zur Anwendung. Im Verlauf Umstellung auf fusidinsäurehaltige Creme. Eine Basistherapie erfolgte im Bereich der betroffenen Areale mittels ureahaltiger Creme. Unter dieser Therapie kam es zu einer Stabilisierung des Hautbefundes mit partieller Lösung der Hyperkeratosen. Bei Kolonisation mit MRSA erfolgte die Einleitung einer dekontaminierenden Therapie mit antiseptischen Waschlösungen. Während des gesamten stationären Aufenthaltes erfolgte die symptomadaptierte Gabe von 02 per Nasensonde.
Kommentar
Die Incontinentia pigmenti stellt eine seltene Genodermatose dar, welche einem X-Chromosomal dominanten Erbgang folgt, eine sehr variable Expressivität besitzt und sich an verschiedenen Organsystemen (Haut, Haare, Nägel, ZNS, Augen, kardiopulmonales System, Immunsystem) manifestieren kann. Nach der ersten umfassenden Beschreibung des Krankheitsbildes durch Bloch (1926) und Sulzberger (1927) wurde der synonyme Begriff des Bloch-Sulzberger-Syndroms geprägt. Das histologisch kennzeichnende Muster der Melanin- und Pigmentinkontinenz im Bereich der betroffenen Areale führte zu dem Eigennamen Incontinentia pigmenti.
Die Erkrankung ist mit circa 1200 weltweit dokumentierten Fällen äußerst selten. Sie betrifft aufgrund des
X-Chromosomal dominanten Erbgangs in der überwiegenden Anzahl weibliche Individuen, da die hemizygote Form bei männlichen Feten meist schon intrauterin letal verläuft. Die zu Grunde liegende genetische Alteration ist eine Mutation im IKBKG-Gen, welches für die Expression des Transkriptionsfaktors NF-kB verantwortlich ist.
Eine komplexe genetische Fehlregulation führt zur mesound ektodermalen Dysplasie. So manifestiert sich die Incontinetia pigmenti bereits kurz nach der Geburt an der Haut, kann aber ebenfalls mit schwerwiegenden Fehlbildungen andere Organsysteme einhergehen. Systemische Beteiligung findet sich bei bis zu 80 % der Betroffenen. Eine Beteiligung des ZNS (z. B. symptomatische Enzephalopathien) oder des kardiopulmonalen Systems (z. B. pulmonaler Hypertonus) limitiert oft die Prognose. Incontinentia pigmenti besitzt eine hohe Penetranz, ist allerdings in der Expressivität äußerst variabel. Daher kann derselbe Genotyp mit sehr unterschiedlichen Phänotypen einhergehen, wie unserem Fallbeispiel von Mutter und Tochter illustriert.
Klinisch zeigt die Dynamik der kutanen Manifestation vier Stadien, wobei nicht alle durchlaufen werden müssen. Die Dauer der Stadien kann intraindividuell verschieden sein, und die Läsionen können im Verlauf komplett abheilen. Das erste Stadium beginnt oft kurz nach der Geburt und ist hauptsächlich durch vesiko-bullöse Effloreszenzen charakterisiert, welche sich entlang der Blaschko-Linien orientieren. Das zweite Stadium geht mit verrukösen, hyperkeratotischen Plaques einher. Im dritten Stadium dominieren streifige, teils wirbelartig konfigu-rierte Hyperpigmentierungen, welche in charakteristischer Weise entlang der Blaschko-Linien ausgerichtet sind. Es schließt sich das vierte Stadium an, das durch lineäre atrophe Hypopigmentierungen und Alopezie, besonders im Bereich der Extremitäten gekennzeichnet ist, und bis in das Erwachsenenalter fortbestehen kann.
Therapeutisch steht bei der Incontinetia pigmenti aus dermatologischer Sicht die Prävention und Kontrolle von potentiellen Superinfektionen der Läsionen besonders im vesikobullösen und verrucösen Stadium im Vordergrund. Eine weitere Option stellt die Applikation von topischen Glukokortikosteroiden zur symptomatischen Therapie dar. Es sollte eine enge interdiziplinäre Kooperation zwischen Dermatologen, Pädiatern und Humangenetikern erfolgen, um sowohl kutane als auch systemische Manifestationen der Erkrankung frühzeitig zu diagnostizieren und zu therapieren sowie eine adäquate humangenetische Beratung der betroffenen Familien zu gewährleisten.
Fazit
Incontinetia pigmenti ist eine im frühsten Kindesalter beginnende seltene Genodermatose, welche eine höchst variable Expressivität aufweist. So kann ein Genotyp, der bei einer Mutter nur geringe kutane Manifestationen bedingt, bei ihren Nachkommen schwerste systemische Manifestationen mit vital bedrohlichen Komplikationen hervorrufen. Charakteristisch ist ein kutaner Verlauf in vier Stadien. Die kutanen Manifestationen einer Incontinentia pigmenti sollten immer auch eine Abklärung der möglichen Beteiligung weiterer Organe veranlassen.
Literatur
Berlin AL, Paller AS, Chan LS et al. (2002) Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol 47:169-187.
Fusco F, Bardaro T, Fimiani G et al. (2004) Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-kappaB activation. Hum Mol Genet 13:1763-1773.
Fusco F, Pescatore A, Bal E et al. (2008) Alterations of the IKBKG locus and diseases: an update and a report of 13 novel mutations. Hum Mutat 29:595-604.
Hayes IM, Varigos G, Upjohn EJ et al. (2005) Unilateral acheiria and fatal primary pulmonary hypertension in a girl with incontinentia pigmenti. Am J Med Genet 135:302-303.
Muhlenstadt E, Eigelshoven S, Hoff NP et al. (2010) Incontinentia pigmenti (Bloch-Sulzberger- Syndrom). Hautarzt 61:831-833.
Phan TA, Wargon O, Turner AM et al. (2005) Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol 30:474-480.