Bei der hereditären kutanen Leiomyomatose (OMIM 150800) findet sich eine Assoziation von mitunter schmerzhaften kutanen und uterinen Leiomyomen. Dieses Syndrom war früher unter dem Namen „Reeds-Syndrom“ bekannt und wird im englischen Sprachgebrauch auch als „multiple cutaneous and uterine leiomyomatosis syndrome“ bezeichnet. Aufgrund möglicher gynäkologischer Komplikationen wird bei betroffenen Patientinnen häufig bereits vor dem 30. Lebensjahr eine Hysterektomie durchgeführt. Zudem besteht ein deutlich erhöhtes Risiko für die Entwicklung von Nierenzellkarzinomen, ein Krankheitsbild, das als hereditäre Leiomyomatose mit Nierenzellkarzinom (OMIM 605839) bezeichnet wird. Beiden Erkrankungen liegen heterozygote Keimbahnmutationen im FH-Gen zugrunde. Dieses Gen kodiert für das gleichnamige Enzym FH, das eine wichtige Rolle im Zitratzyklus einnimmt und die Konvertierung von Fumarat zu Malat katalysiert. Homozygote FH-Mutationen führen bereits früh zu schwersten Entwicklungsstörungen und sind in der Regel nicht mit dem Leben vereinbar. Mittlerweile konnten bereits über 70 verschiedene FH-Mutationen nachgewiesen werden.
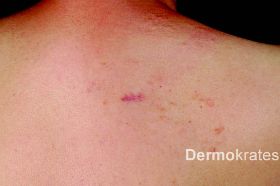
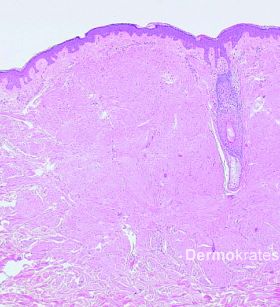
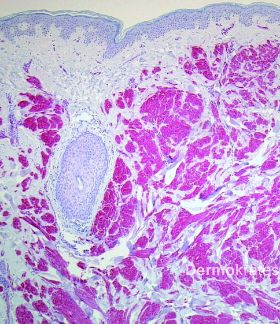
Kutane Leiomyome, auch als Piloleiomyome bezeichnet, sind seltene, glattmuskulär differenzierte Tumoren der Haut. Sie zeigen ein sehr variables klinisches Erscheinungsbild und können als solitäre Läsionen oder disseminiert auftreten. Wichtigste Differentialdiagnosen sind Mastozytome, eruptive Syringome, Neurofibrome, Trichofollikulome oder papulöse Akneläsionen. Da bei der hereditären Leiomyomatose häufig zuerst die kutanen Leiomyome auftreten, stellen sie eine wichtige Markerläsion zur frühzeitigen Diagnose assoziierter Organbeteiligungen dar und sollten daher möglichst schnell durch eine histologische Untersuchung verifiziert werden.
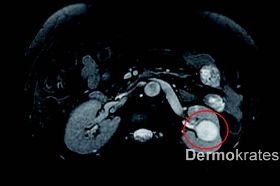
Die derzeitigen Behandlungsmöglichkeiten kutaner Leiomyome sind leider beschränkt. Operative Maßnahmen oder eine analgetisch-symptomatische Therapie mit z.B. Gabapentin oder Nifedipin sind therapeutische Optionen. Betroffene Frauen sollten zudem regelmäßig gynäkologisch untersucht werden und bei Vorliegen von Uterusmyomen bereits frühzeitig eine Beratung im Hinblick auf die Familienplanung erhalten. Die wichtigste Maßnahme bei hereditärer kutaner Leiomyomatose ist jedoch ein jährliches Screening zur Früherkennung eines Nierenzellkarzinoms. Wie am Beispiel unserer Patientin deutlich wird, ist die alleinige Durchführung einer Sonographie jedoch nicht ausreichend, da aufgrund einer möglichen identischen Dichte von Tumorgewebe und Nierenparenchym durch diese Untersuchung allein keine ausreichende Sensitivität gegeben ist. In der Regel wird daher die Durchführung eines CT-Abdomens oder eine MRT-Untersuchung empfohlen, wobei es derzeit noch keine allgemein anerkannten Leitlinien gibt.
Eine FH-Mutationsanalyse zur Sicherung der Diagnose wie bei unserer Patientin ist in spezialisierten Zentren möglich und sollte immer zusammen mit einer humangenetischen Beratung erfolgen, um den Betroffenen die Konsequenzen der Diagnose nahezubringen, betroffene Familienmitglieder zu identifizieren und ebenfalls in ein Screening-Programm integrieren zu können.