Birt-Hogg-Dubé-Syndrom
-
Schwierigkeitsgrad Fortgeschrittene
42 Faziale Papeln und Pneumothoraces – Birt-Hogg-Dubé-Syndrom
Last Updated: 12-08-2013
Verena Lichte¹, Julia Reifenberger¹, Michel van Geel³, Jorge Frank¹,²
1 Hautklinik und 2 Hauttumorzentrum, Universitätsklinikum Düsseldorf
2 Afdeling Dermatologie; Maastricht Universitair Medisch Centrum (MUMC), Nederland
vorgestellt als Dia-Klinik, 46. Tagung der Deutschen Dermatologischen Gesellschaft, Dresden 2013
DOI
10.1111/ddg.12061
Anamnese
In unserer Ambulanz sahen wir drei Patienten, bei denen sich in der vierten Lebensdekade Hautveränderungen im Gesicht manifestiert hatten.
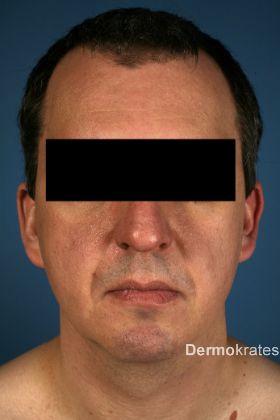
Der erste Patient, ein 43-jähriger Mann kaukasischer Abstammung, stellte sich mit seit drei Jahren bestehenden und sich ausbreitenden, zwei bis drei Millimeter großen weißlich-hautfarbenen Papeln im Gesichts- und Halsbereich vor (Abb. 1a). Er berichtete über rezidivierende Pneumothoraces in den 1990er Jahren. Bei Vater und Schwester bestünden ähnliche Hautveränderungen.
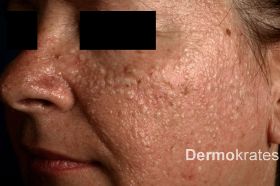
Die zweite Patientin, eine 51-jährige kaukasische Frau, berichtete über seit 13 Jahren zunehmende, teils juckende, ebenfalls zwei bis drei Millimetern große weißlich-hautfarbene Papeln im Gesicht, am Kopf, Hals und Nacken (Abb. 1b). Daneben wies die Anamnese mehrere Spontanpneumothoraces, Nierenparenchymzysten und ein uterines Leiomyom auf.
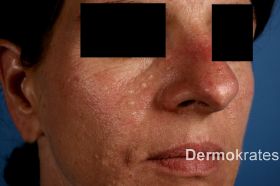
Die dritte Patientin, eine 42-jährige Kaukasierin, berichtete über seit acht Jahren progrediente weißlich-hautfarbene Papeln von zwei bis drei Millimetern im Gesicht und Décolleté (Abb. 1c). Die Eigen- und Familienanamnese war unauffällig.
Bezüglich des weiteren Hautbefundes der drei Patienten waren die Mundschleimhaut und das übrige Integument nicht betroffen.
Histopathologie
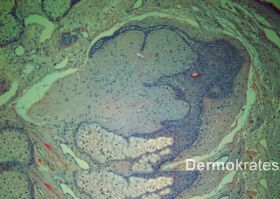
In einer Probebiopsie einer fazialen Papel von Patientin 2 stellten sich unter einer papulös vorgewölbte ortholog strukturierte Epidermis an umschriebener Stelle im oberen Korium Proliferationen von epithelialen Zellstängen dar, die keine Verbindung zur Epidermis erkennen ließen. Diese waren nur wenige Zelllagen breit und konfluierten zum Teil in der Tiefe. An einer Stelle kam ein rudimentärer Haarfollikel zur Darstellung sowie Anteile einer Talgdrüse. Umgeben waren die epithelialen Proliferate von lockerem Bindegewebe mit Nachweis von Muzin und spindeligen Fibroblasten im Sinne eines Fibrofolliuloms (Abb. 2).
Bildgebende Diagnostik
Beim ersten Patienten zeigten sich in der Magnetresonanztomographie (MRT) beide Nieren regelrecht. Während halbjährliche sonographische und eine initiale MRT-Untersuchung bei der zweiten Patientin Nierenparenchymzysten ergab, waren diese Untersuchungen bei der dritten Patientin unauffällig.
Molekulargenetische Analyse
In der DNA-Sequenzanalyse aller kodierenden Abschnitte des Folliculin (FLCN)-Gens fand sich beim ersten Patienten in Exon 5 eine heterozygote Mutation mit der Designation c.256_269del14.
Therapie und Verlauf
Allgemein
Nach Diagnosestellung und humangenetischer Beratung führten wir beim ersten Patienten an Stirn und Hals eine Behandlung mittels Erbium:YAG-Laser in mehreren Sitzungen durch. Die zweite Patientin wünschte lediglich eine Diagnosestellung und genetische sowie kosmetische Beratung, jedoch keine Therapie. Hingegen nahmen wir bei der dritten Patientin eine Probelaserung von sieben Papeln an der Wange mit dem Erbium:YAG-Laser vor. Bei beiden behandelten Patienten führte die Lasertherapie zu einem zufriedenstellenden ästhetischen Ergebnis.
Kommentar
In Zusammenschau der erhobenen Befunde stellten wir bei allen Patienten die Diagnose Birt-Hogg-Dubé-Syndrom (BHDS), das durch die familiär gehäufte Manifestation von Fibrofollikulomen, Trichodiskomen und Akrochorda in Assoziation mit Lungenzysten, spontan auftretenden Pneumothoraces sowie benignen und malignen Nierentumoren charakterisiert ist. Dieses Tumorsyndrom hat eine geschätzte Prävalenz von 1:200.000 und gehört somit zu den seltenen Erkrankungen.
An der Haut manifestieren sich üblicherweise zwischen dem 30. und 40. Lebensjahr wenige Millimeter große, weißlich-hautfarbene Papeln, insbesondere an Wangen, Nase, Hals und Kopfhaut, teilweise auch an Mundschleimhaut und Oberkörper. Das klinische Erscheinungsbild kann von wenigen solitären Papeln bis hin zum großflächigen Befall variieren. Die in der Regel erst im späteren Verlauf der Erkrankung auftretenden renalen Veränderungen beinhalten benigne Nierenzysten, Onkozytome, maligne chromophobe, klarzellige oder papilläre Nierenzellkarzinome und gemischte Nierentumoren.
Die Diagnose des BHDS basiert auf der frühzeitigen Erkennung der klinischen Manifestationen, einer positiven Familienanamnese und der histopathologischen Untersuchung. Die endgültige Bestätigung kann durch eine molekulargenetische DNA-Analyse erbracht werden. Das BHDS wird autosomal dominant vererbt. Ursächlich sind heterozygote Mutationen im FLCN-Gen, das auf Chromosom 17p11.2 liegt und für das gleichnamige Protein Folliculin kodiert, dessen Funktion weitestgehend unbekannt ist. Vermutet wird, dass Folliculin eine wichtige Rolle in der mTOR ("mammalian target of rapamycin")-Signalroute spielt. Einerseits könnten Mutationen im FLCN-Gen zur Dysregulation dieses Signalweges führen und somit zur Entwicklung von Nierentumoren und anderen assoziierten Organveränderungen beitragen. Andererseits könnte FLCN die Funktion eines Tumorsuppressor-Gens haben, so dass durch FLCN-Keimbahnmutationen die Kontrollfunktion von Folliculin auf das Zellwachstum gestört und hierdurch die Entstehung von Nierentumoren begünstigt wird.
Wie beim ersten Patienten steht heutzutage bei Individuen mit multiplen kutanen Papeln im Sinne von
Fibrofollikulomen oder Trichodiskomen mit der molekulargenetischen Analyse ein diagnostisches Verfahren zur Verfügung, das bei unspezifischen klinischen oder histologischen Befunden das BHDS zweifelsfrei verifizieren kann. Nach Bestätigung der Diagnose sollten aufgrund der Assoziation mit Nierenzellkarzinomen ab dem 20. Lebensjahr eine interdisziplinäre Betreuung und regelmäßige Vorsorgeuntersuchungen mittels halbjährlicher sonographischer und/oder jährlicher MRT eingeleitet werden. Bei den hier vorgestellten Patienten konnte dadurch die Manifestation maligner Nierentumore bislang ausgeschlossen werden.
Derzeit gibt es keine spezifischen Therapieleitlinien hinsichtlich der dermatologischen Manifestationen des BHDS. Solitäre Tumoren können chirurgisch entfernt werden, wobei auch die Dermabrasion, Elektro-Desikkation oder ablative Lasertherapie zur Anwendung kommen können.
Zusammenfassend müssen multiple faziale Papeln in Zusammenhang mit rezidivierenden Pneumothoraces als Hinweis auf ein BHDS gesehen werden. In Bezug auf mögliche Assoziation mit Nierenzellkarzinomen sind regelmäßige Screening-Untersuchungen und eine interdisziplinäre Betreuung der Patienten unabdingbar.
Literatur
1 Birt AR, Hogg GR, Dube WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977; 113: 1674-1677.
Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, Nordenskjöld M, Hansen TV, 2 Solly J, Maher ER. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 2009; 10: 1199-1206.
3 Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S, Pavlovich CP, Glenn G, Greenberg CR, Linehan WM, Zbar B. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001; 69: 876-882.
4 van Steensel MA, Verstraeten VL, Frank J, Kelleners-Smeets NW, Poblete-Gutiérrez P, Marcus-Soekarman D, Bladergroen RS, Steijlen PM, van Geel M. Novel mutations in the BHD gene and absence of loss of heterozygosity in fibrofolliculomas of Birt-Hogg-Dubé patients. J Invest Dermatol 2007; 127: 588-593.
5 Warren MB, Torres-Cabala CA, Turner ML, Merino MJ, Matrosova VY, Nickerson ML, Ma W, Linehan WM, Zbar B, Schmidt LS. Expression of Birt-Hogg-Dubé gene mRNA in normal and neoplastic human tissues. Mod Pathol 2004; 17: 998-1011.