Erythropoetische Protoporphyrie
ICD-11: AIB2
-
Schwierigkeitsgrad Fortgeschrittene
52 Erstdiagnose einer erythropoetischen Protoporphyrie bei einem Geschwisterpaar ohne Familienanamnese
Last Updated: 12-08-2013
Michael Erdmann, Florian Anzengruber, Miklós Simon jr.
Hautklinik Erlangen, Ulmenweg 18, 91052 Erlangen
vorgestellt als Dia-Klinik, 46. Tagung der Deutschen Dermatologischen Gesellschaft, Dresden 2013
DOI
10.1111/ddg.12061
Anamnese
Im Juni 2012 stellte sich ein 17-jähriger Patient mit scharf auf lichtexponierte Areale beschränkten Ödemen, Rötungen, punktförmigen Einblutungen und umschriebenen Erosionen ohne Krankheitsgefühl oder aktuelle Medikamenteneinnahme vor. Am Vortag hielt er sich mittags ca. 30 Minuten mit T-Shirt im Freien auf. Auf Kindheitsfotos kam es wiederholt ab halbstündiger direkter Sonnenbestrahlung innerhalb weniger Stunden an lichtexponierter Haut zu Ödemen, Spannung und Rötung gefolgt von Einblutung und Blasenbildung. In der Familie entwickelte nur die Schwester derartige Hautveränderungen nach Lichtexposition.
Hautbefund
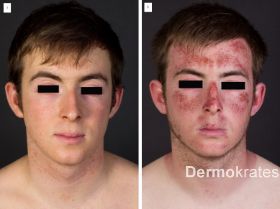
Bevorzugt auf Wangen und Nasenrücken beschränkte diskrete Rötung und vereinzelte Narben (Abb. 1a). Nach Lichtexposition scharf auf die belichteten Hautareale begrenzte ausgeprägte Ödeme, Erytheme, Petechien im Gesicht und Hals sowie Erosionen an Ohrhelices und Nacken (Abb. 1b).
Befunde
Patient
Erhöht: Leukozyten 11,4 x 103/μl (4,0-10,0), Erythrozyten 6,15 x 106/μl (4,2-5,9), Monozyten 12 % (2-8), Bilirubin 1,3mg/dl (<1,1), ALT 75 U/l (<50), LDH 256 U/l (105-235), C-reaktives Protein 90,5 mg/dl (<5).
Erniedrigt: Lymphozyten 13 % (25-50),
Normwertig: Gerinnung und antinukleäre Antikörper.
Porphyrindiagnostik:
Plasmafluoreszenz-Scan (580-650nm): positiv, Emissionswellenlänge 624 nm
Freies Protoporphyrin (Erythrozyten): 19314 nmol/l (9,0-89,0)
Zink-Protoporphyrin: 117,0 μmol/mol (<40,0)
Gesamt-Protoporphyrin (Erythrozyten): 20302 nmol/l (<540)
Kein Material zur Urin-, Stuhluntersuchung.
Oberbauchsonographie: Keine Cholezystolithiasis oder fokale Leberläsion.
Schwester
Plasmafluoreszenz-Scan (580-650nm): positiv, Emissionswellenlänge 624 nm
Freies Protoporphyrin (Erythrozyten): 14337 nmol/l (9,0-89,0)
Zink-Protoporphyrin 96,7 μmol/mol (<40,0)
Gesamt-Protoporphyrin (Erythrozyten) 15105 nmol/l (<540)
Oberbauchsonographie: Keine Cholezystolithiasis oder fokale Leberläsion.
Vater
Freies Protoporphyrin (Erythrozyten): 288,5nmol/l (9,0-89,0)
Therapie und Verlauf
Allgemein
Bis zur Wiedervorstellung im Oktober 2012 kam es bei dem Geschwisterpaar unter konsequentem Lichtschutz, Meiden von längerer Lichtexposition sowie oral Beta-Carotin 75 mg täglich und Vitamin-D-Supplementierung zu keinen erneuten Hautveränderungen im Rahmen der erythropoetischen
Protoporphyrie. In der Humangenetischen Untersuchung wurde bei beiden Geschwistern Mutationen im FECH-Gen (Ferrochelatase) nachgewiesen.
Kommentar
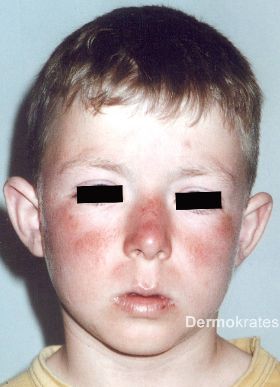
Der erythropoetischen Protoporphyrie liegt eine genetisch verminderte Aktivität der Ferrochelatase (Chromosom 18q21.3) in der Hämbiosynthese zugrunde. Die Erkrankung hat eine Prävalenz von 0,5-1 pro 100.000 Einwohner. Wie bei unseren Patienten erfolgt häufig ein sogenannter pseudodominanter Erbgang: Ein Allel weist geringe bis fehlende Aktivität (mehr als 135 beschriebene Mutationen), das andere, normale Allel eine niedrige Genexpression der Ferrochelatase auf. Bei erhöhtem freien Protoporphyrin im Blut des Vaters, ist dieser der wahrscheinliche Träger der Mutation mit geringer bis fehlender Aktivität. Bei mehr als um 50 % reduzierter Ferrochelatase-Aktivität kommt es zu erhöhtem freien Protoporphyrin, das sich als [1] akute kutane UVA-Photosensitivität bereits im jungen Kindesalter (Abb. 2) mit Jucken, Brennen, Stechen, Erythemen, Ödemen, Purpura, Blasen, Erosionen sowie Narben und [2] hepatobilär als Leberschädigung bis zum akuten Leberversagen in 2-5 % der Patienten und Gallensteinen manifestiert.
Differenzialdiagnostisch kommen weitere lichtprovozierbare Dermatosen wie andere Formen der kutanen Porphyrien, eine medikamentös induzierte Photosensitivität, eine Lichturtikaria, ein systemischer Lupus erythematodes oder eine polymorphe Lichtdermatose infrage.
Die Therapie besteht aus konsequentem Lichtschutz und zusätzlich Einnahme von Beta-Carotin und Vitamin D/Ascorbinsäure mit unterschiedlichem Erfolg. In aktuellen Phase-3-Studien wird bei der erythropoetischen Protoporphyrie subkutan Afamelanotide, ein Alpha-MSH-Analogon, verabreicht. Hierunter kommt es zu einer verstärkten Pigmentierung der Haut, die zu einer Steigerung der Schmerztoleranz und verlängerter Expositionsdauer gegenüber Sonnenlicht führt. Bei akutem Leberversagen kommt eine Lebertransplantation in Betracht.
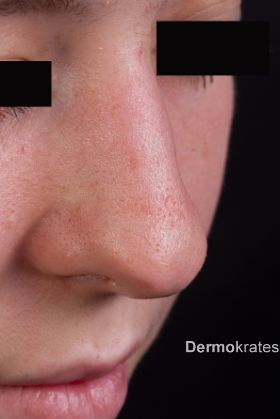
Aufgrund der typischen Anamnese mit ausgeprägter Lichtempfindlichkeit wird die Diagnose einer erythropoetischen Protoporphyrie meist im jungen Kindesalter gestellt. Bei Vorstellung von Patienten ohne akuten Schub sind kleine auf den ersten Blick unscheinbare Narben an Hand-/Finger-/Nasenrücken wie bei der Schwester vorliegend (Abb. 3) klinisch wegweisend.
Danksagung
Wir danken Herrn PD Dr. med. RA Jamra, Humangenetisches Institut, Universitätsklinikum Erlangen für seine genetischen Untersuchungen.
Literatur
1 Günther H. Die Hämatoporphyrie. Dtsch Arch Klein Med 1911; 105: 89-145.
2 Harms J, Lautenschlager S, Minder CE, Minder EI. An alphamelanocyte-stimulating hormone analogue in erythropoetic protoporphyria. N Engl J Med 2009; 360: 306-307.
3 Hönigsmann H. Die erythropoetische Protoporphyrie I. Klinik und Problematik der Pathogenese. Z Hautkr 1977; 52: 485-509.
4 Kosenow W, Treibs A. Lichtüberempfindlichkeit und Porphyrinämie. Z. Kinderkrankh 1953; 73: 82-92.
5 Langhof H. Untersuchung zur familiären, protoporphyrinämischen Lichturticaria. Archiv für Klinische und Experimentelle Dermatologie 1961; 212: 506-518.
6 Magnus IA, Jarret A, Pankered TAJ et al. Erythropoetic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinemia. Lancet 1961; 2: 448-451.