Bereits bei Geburt (sekundäre Sectio caesarea bei pathologischem CTG) fielen bei dem nach unkomplizierter Schwangerschaft reifgeborenen Mädchen (40+2 SSW, Gewicht: 3430 g, Länge 53 cm, Kopfumfang 34 cm) livide, netzartige, nicht schmerzhafte Erytheme auf. Keine Schleimhautblutungen. Der Säugling war zu jedem Zeitpunkt kardiorespiratorisch stabil und ohne klinische Hinweise auf eine Infektion (Apgar 9/10/10 nach 1/5/10 Minuten). Mit der Verdachtsdiagnose einer generalisierten Purpura erfolgte wenige Stunden postnatal die Aufnahme auf die Neugeborenenintensivstation.
-
Schwierigkeitsgrad Fortgeschrittene
9 Cutis marmorata teleangiectatica congenita
Rita Varga, Ilana Goldscheider, Sarah Basedow, Kathrin Giehl, Peter Kaudewitz
Klinik und Poliklinik für Dermatologie und Allergologie der Ludwig-Maximilians-Universität München
vorgestellt als Dia-Klinik, 23. Fortbildungswoche für praktische Dermatologie und Venerologie, München 2012
DOI
Hautbefund
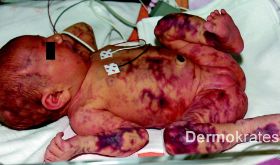
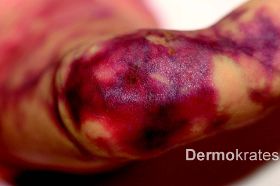
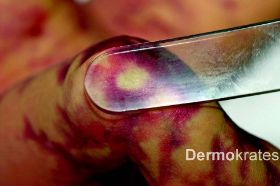
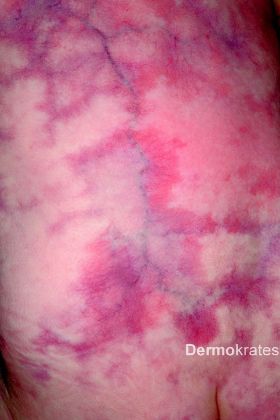

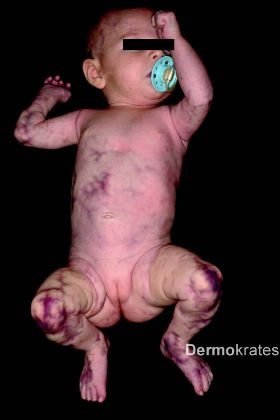
Generalisiert an Stamm, Extremitäten und Kopf mit Aussparung des Gesichts bizarre bläulich-rote, asymmetrische, teils netzartige, mit dem Diaskop wegdrückbare Erytheme. Daneben Teleangiektasien, vereinzelt prominente Venen und stellenweise, insbesondere an den Knien, Hautatrophie und hypotrophes subkutanes Fettgewebe. Der Gesamtaspekt erinnert an Marmor.
Histopathologie
Gluteal: korbgeflechtartige Orthokeratose sowie kompaktes und schmales Stratum granulosum. Das Epidermisband ist schmal, keine ausgeprägte Reteleistenarchitektur. Subepidermal erweiterte Gefäßlumina. Im kollagenen Gewebe diskretes lymphohistiozytäres Infiltrat.
Beurteilung: Befund vereinbar mit Cutis marmorata teleangiectatica congenita.
Labor
Klinische Chemie: Bilirubin direkt mit 0,6 mg/dl [<0,3] erhöht, sonst unauffällig.
Hämatologie: Lymphozyten mit 15 % [18-65] erniedrigt, Segmentierte mit 76% [15-60] erhöht, Anisozytose, Polychromasie, sonst unauffällig.
Immunologie: Doppelstrang-DNS-Ak, Cardiolipin-IgG-Ak, Cardiolipin-IgM-Ak, β2-Glykoprotein I IgG-Ak und β2-Glykoprotein I IgM-Ak ohne pathologischen Befund.
Neugeborenenscreening einschließlich TSH-Test: unauffällig.
Apparative Diagnostik
EKG: Sinusrhythmus, Steil-/Indifferenztyp, normale Herzzeitmaße, keine Erregungsrückbildungsstörung, keine Hypertrophiezeichen.
Echokardiographie: Kleiner muskulärer drucktrennender Ventrikelseptumdefekt, Kontrolle empfohlen.
Sonographie Abdomen und Schädel: kein pathologischer Befund.
Röntgen des Thorax: kein pathologischer Befund.
Otoakustische Emissionen: beidseits unauffällig.
Augenärztliches Konsil: kein Anhalt für Beteiligung der Augen. Kein Anhalt für Glaukom. Kontrolle in 6 Monaten empfohlen, bei Trübung der Hornhaut früher.
Therapie und Verlauf
Allgemein
Im Bereich der atrophen Areale an den Knien bildeten sich Wunden, die mit Verbandswatte abgepolstert wurden. Es lagen keine Ulcera vor. Bei der dermatologischen Kontrolluntersuchung drei Monate postnatal fiel eine deutliche Besserung des Hautbefundes mit Rückgang und Abblassen der betroffenen Areale auf. Zu dem Zeitpunkt waren keine weiteren krankheitsassoziierten Anomalien zu verzeichnen.
Kommentar
Die Cutis marmorata teleangiectatica congenita (CMTC) wurde erstmals 1922 durch die niederländische Kinderärztin van Lohuizen beschrieben. Es handelt es sich um eine angeborene, meist sporadisch auftretende Erkrankung, die in der Regel bereits bei Geburt vorhanden ist. Selten tritt die CMTC erst nach der Geburt innerhalb der ersten zwei Lebensjahre auf. Hauptmerkmal der CMTC ist eine durch Teleangiektasien und Phlebektasien hervorgerufene netzförmige Zeichnung der Haut. Die Gefäßmuster sind meist bizzar, von rötlich-livider Farbe, in der Regel wegdrückbar, und treten beim Wechsel von warm nach kalt stärker hervor. Zumeist tritt die CMTC segmental auf, sie kann aber auch in diffuser Weise nahezu das gesamte Integument betreffen. Nicht selten sind Hautatrophien im Bereich der Läsionen und lokalisierte Atrophien des subkutanen Fettgewebes sowie Ulzerationen vorhanden. Die betroffene Haut erscheint transparent und marmoriert. Die histopathologischen Befunde mit erweiterten Kapillaren und Venolen in der Dermis und Subkutis sind oft unspezifisch. Sie können die Diagnose nicht eindeutig sichern und tragen nicht wesentlich zur Abgrenzung anderer Differenzialdiagnosen bei, weshalb eine Hautbiopsie meist entbehrlich ist.
Mehr als 50 % der CMTC-Patienten haben assoziierte kutane und/oder extrakutane Anomalien. Häufig sind dabei Körperasymmetrie (Hypo- oder Hypertrophie einer Extremität), Veränderungen am Skelettsystem, Augenanomalien (Netzhautablösung und kongenitales Glaukom) oder kapilläre Fehlbildungen (z. B. Naevus flammeus). Weniger häufig finden sich Gefäßanomalien (z. B. zerebrovaskuläre Malformationen), Auffälligkeiten des Schädels (z. B. Makrozephalie) und Zentralnervensystems (z. B. mentale Retardierung), Anomalien des Bewegungsapparates (z.B. Spina bifida) oder auch genitoanale Missbildungen (z. B. Klitorisaplasie).
Die Ursache der Erkrankung ist nach wie vor nicht geklärt. Diskutierte Hypothesen gehen von einer autosomal-dominanten Vererbung mit variabler Penetranz oder einem Mosaik eines sonst letalen, bisher unbekannten autosomal dominanten Gendefekts aus. Exogene teratogene Faktoren werden ebenfalls postuliert.
Die Diagnose basiert auf der charakteristischen Klinik. Sie sollte insbesondere im Hinblick auf assoziierte Anomalien möglichst frühzeitig erfolgen. Die apparative Diagnostik sollte zeitnah ophthalmologisch z. A. eines Glaukoms, neuroradiologisch z. A. von Hirnfehlbildungen und orthopädisch/radiologisch zur Klassifizierung von Skelettfehlbildungen herangezogen werden. Differenzialdiagnostisch müssen die physiologische Livedo reticularis des Neugeborenen, die Livedo racemosa, der neonatale Lupus erythematodes, das Klippel-Trenaunay-Syndrom und das Sturge-Weber-Syndrom berücksichtigt werden. Liegt eine Kombination der CMTC mit Extremitäten-, Schädelfehlbildungen und Störungen des ZNS vor, kommt ein Adams-Oliver-Syndrom als mögliche Maximalvariante in Betracht.
Kausale Behandlungsmöglichkeiten bei CMTC fehlen bislang. Eine Therapie der dermatologischen Manifestationen ist meist nicht notwendig. Die symptomatische Behandlung beinhaltet Kälteschutz und Krankengymnastik, insbesondere bei Körperasymmetrie. Die Eltern sollten über den relativ günstigen Verlauf mit häufig spontaner Rückbildungstendenz (>50 %) in den ersten beiden Lebensjahren aufgeklärt werden. Persistierende Hautläsionen können ab einem Alter von etwa zehn Jahren mit Lasern behandelt werden, wobei Schmerzhaftigkeit und fehlende Erfolgsgarantie limitierende Faktoren darstellen. Zur Abklärung assoziierter Fehlbildungen sind bei jedem Patienten regelmäßig neben kinderärztlichen und dermatologischen auch spezielle augenärztliche, kinderneurologische und orthopädische Kontrolluntersuchungen vorzunehmen.
Fazit
CTMC ist eine seltene, konnatale Gefäßanomalie, die durch marmorartige, bizarr-livide Hautverfärbungen sowie Phlebektasien, Teleangiektasien, Atrophien und gelegentlich Ulzerationen gekennzeichnet ist. Die Erkrankung zeigt häufig eine spontane Rückbildungstendenz (>50 %) in den ersten Lebensjahren. Betroffene Patienten benötigen eine fächerübergreifende Langzeitüberwachung um assoziierte Anomalien (Körperasymmetrie, Glaukom, ZNS-Störungen) frühzeitig behandeln zu können.
Literatur
Garzon MC, Schweiger E (2004) Cutis marmorata teleangiectatica congenital. Semin Cutan Med Surg 23: 99-106
Kienast AK, Hoeger PH (2009) Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol 34: 319-323.
Picascia DD, Esterly NB (1989) Cutis marmorata telangiectatica congenita: report of 22 cases. J Am Acad Dermatol 20: 1098-1104.
Rupprecht R, Hundeiker M (1997) Cutis marmorata teleangiectatica congenita. Wichtige Aspekte für die dermatologische Praxis. Hautarzt 48: 21-25.
Van Lohuizen CHJ (1922) Cutis marmorata telangiectatica congenita. Acta Derm Venereol 3: 202-211.