Angiokeratoma corporis diffusum (ACD)
-
Schwierigkeitsgrad Fortgeschrittene
46 Angiokeratoma corporis diffusum (ACD) ohne Defekt im α-Galaktosidase-Gen
Last Updated: 20-08-2013
Heike Schulenburg¹, Annette Stein¹, Gustavo Bruno Baretton², Stefan Beissert¹
1 Universitätsklinikum Carl-Gustav-Carus, Klinik und Poliklinik für Dermatologie, TU Dresden
2 Universitätsklinikum Carl-Gustav-Carus, Institut für Pathologie, TU Dresden
vorgestellt als Dia-Klinik, 46. Tagung der Deutschen Dermatologischen Gesellschaft, Dresden 2013
DOI
10.1111/ddg.12061
Anamnese
Ein aktuell 64-jähriger Mann stellte sich mit seit dem 30. Lebensjahr zunehmenden, zum Teil rupturierenden, Angiokeratomen am gesamten Integument vor. Der ältere der zwei Söhne (34-jährig) zeige aktuell beginnend ebenfalls gefäßreiche Hautveränderungen. Der Hautbefund der Eltern sei unauffällig, die Mutter erlitt zwei zerebrale Ischämien.
Hautbefund
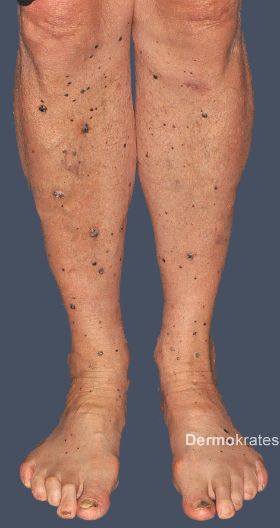
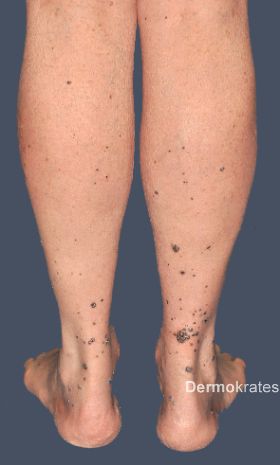
Bei unserem Patienten zeigen sich am gesamten Integument, mit Betonung der distalen unteren Extremität und Aussparung des Gesichts, multiple, isolierte, bis maximal 25 x 15 mm messende, dunkelrote bis schwarze, gefäßkonvolutreiche, weiche Nodi. Diese rupturieren zum Teil durch alltägliche mechanische Reizung (z. B. Reibung durch Kleidung) mit anschließender Blutung (Abb. 1a und b).
Dermatohistopathologie
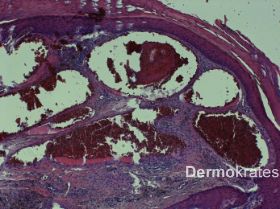
Lakunenartig dilatierte Papillarkörpergefäße mit stellenweiser zytoplasmatischer Vakuolisierung von Endothelienzellen. Hyperkeratotische Hyperplasie im Bereich der erheblich vorgewölbten, bedeckenden Epidermis. Zwischen den stark erweiterten Gefäßen ist reichlich Eisenpigment nachzuweisen. Befund eines Angiokeratoms im Rahmen eines Angiokeratoma corporis diffusum (ACD) (Abb. 2).
Elektronenmikroskopie
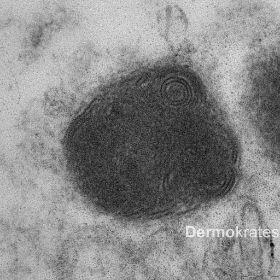
Ultrastrukturell sieht man in einzelnen Endothelien Lysosomen mit konzentrisch geschichteten osmiophilen
Lamellen, vereinzelt auch parallel angeordneten Lamellen. Vielfach sind diese intralysosomalen lamellären Strukturen hochgradig verdichtet, sodass sie nicht mehr voneinander zu trennen sind. Zusammen mit Klinik und Nachweis von lysosomalen Strukturen ist der Befund hinreichend verdächtig auf das Vorliegen eines Morbus Fabry (Abb. 3).
Diagnostik
Neben den histologisch gesicherten Angiokeratomen zeigten sich Begleitsymptome neuronal (multiple
intrazerebrale Kavernome, symptomatische fokale Epilepsie mit Z. n. Grand-Mal, Z. n. lakunärem Infarkt
der Pons mit konsekutiver Gleichgewichtsstörung; keine kutane Schmerzsymptomatik), kardial (arterielle Hypertonie, Sinustachykardie), pulmonal (COPD °II), nephrologisch (Niereninsuffizienz °II) und im Stoffwechsel (Hypothyreose, Dyslipoproteinämie, Hyper-Homocysteinämie). Die Gen-Sequenzierung wies keine α-Galaktosidase(GAL)-Mutation auf.
Therapie und Verlauf
Allgemein
Die Therapie erfolgt rein symptomatisch mit der Exzision großer Angiokeratome. Eine therapeutische Alternative kleinerer Läsionen stellt die Lasertherapie dar.
Kommentar
Diffuse Angiokeratome sind selten, meist assoziiert in Syndromen, ihre Entstehung wird auf Enzymdefekte zurückgeführt, die Therapie ist symptomatisch.
Die Genanlyse zeigte keinen Hinweis auf das Vorliegen eines M. Fabry, des häufigsten X-chromosomal vererbten Enzymdefekts mit Assoziation zu diffusen Angiokeratomen. Differenzialdiagnostisch kommen seltenere Gendefekte oder das Vorliegen ideopatischer diffuser Angiokeratome in Betracht. Bei normaler geistiger und körperlicher Entwicklung unseres Patienten (von Beruf Universitätsprofessor) wird klinisch aktuell ein bekannter Gendefekt ausgeschlossen und von einer ideopatischen Form des Angiokeratoma corporis diffusum ausgegangen. Die Hautveränderungen des Sohns geben jedoch einen Hinweis auf eine genetische Komponente.
Die frühe Diagnosestellung ist wegen erhöhter Inzidenz tödlich verlaufender zerebraler Ischämien sowie anderer Organpathologien wichtig. Präparate zur Substitution fehlerhafter oder mangelnder Enzyme zeigen bei Probanden mit bekanntem Gendefekt eine Reduktion der Symptome. Zu deren Anwendung ist die Kenntnis des genauen Enzymdefekts essentiell.
Literatur
1 Keating GM. Agalsidase alfa: a review of its use in the management of Fabry disease. BioDrugs. 2012; 26(5): 335-54.
2 Larralde MM. [Fabry disease; skin in nutritional, metabolic and heritable disease] Fitzpatrick, McGraw Hill, New York 2008; 136(24): 1281-1288.
3 Schaefer RM, Tylki-Szymańska A, Hilz MJ. Enzyme replacement therapy for Fabry disease: a systematic review of available evidence. Drugs. 2009; 69(16): 2179-205.
4 Sivley MD. Fabry Disease: A Review of Ophthalmic and Systemic Manifestations. Optom Vis Sci. 2013 Jan 17. [Epub ahead of print]